Proteomics Core
Introduction to Proteomics
The proteome refers to the total number of proteins present within either a cell, tissue, or organism at a given time. This protein population can differ over time, between growth conditions, and between cell types, due to differences in gene expression. Proteomics assess the activities, modifications, and interactions within protein complexes, ultimately determining their function. Proteins are intrinsically important within cell systems; maintaining metabolic processes, providing structural elements, and are integral in gene expression acting as signal receptors or initiators and compose the resulting products.
Mass spectrometry is a common tool for proteome analysis. Usually coupled with a liquid chromatography system, this method for protein identification relies on fragment detection and measurement, which when compared to large scale data bases can accurately identify peptide sequences present in samples.
A standard proteomics workflow includes protein extraction, enzymatic digestion, HPLC separation, analysis of the resulting peptides with tandem mass spectrometry (LC-MS/MS), and then database searching or software-based protein quantification. Additional steps can be added to either enrich for specific components or further fractionate the samples.
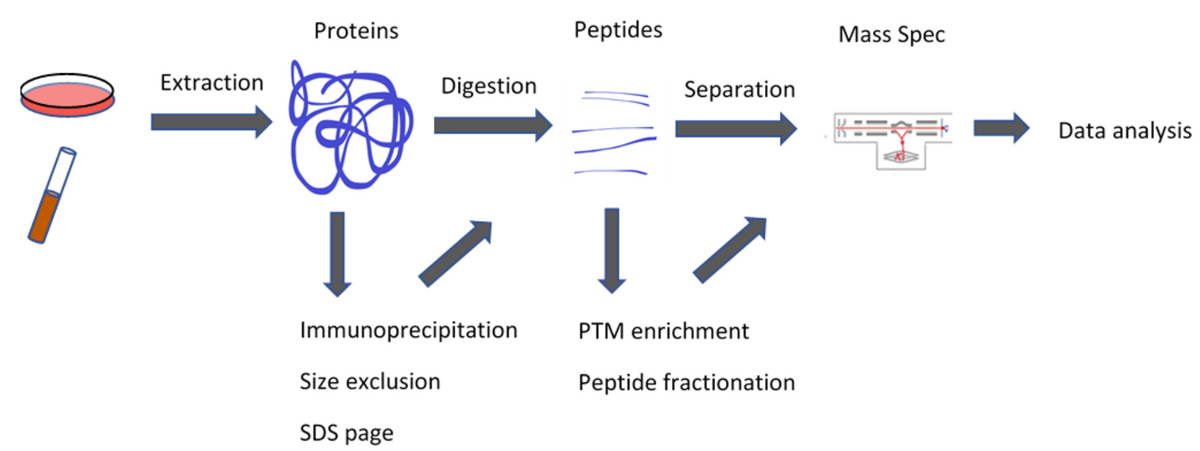
Basic Proteomics Workflow
Mass Spectrometer Data Acquisition: Peptide solutions are run on a gradient for separation. The mass spectrometer is programed to scan for a predetermined mass range (usually from 500-1800 daltons). The highest mass peaks from these LC-MS scans are then isolated and tandem MSMS is performed, fragmenting the peptides and creating an ion pattern. From this electrospray pattern the Δ mass can be determined and both the sequence of amino acids and any post translational modifications can be identified from the original peptide.
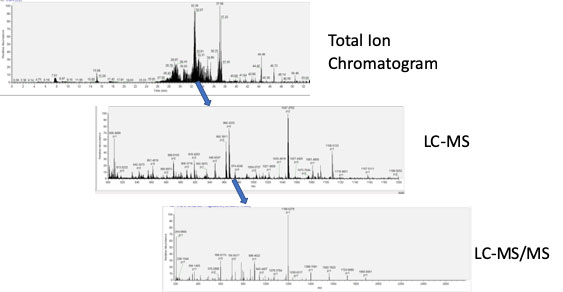
For a quick introduction please read:
Malmstom J., Lee H, Aebersold R. Advances in proteomic workflows for systems Biology. Curr Opin Biotechnol. 2007 18(4): 378-84.
Chen Y, Vu J, Thompson MG, Sharpless WA, Chan LJG, Gin JW, et al. (2019) A rapid methods development workflow for high throughput quantitative proteomic applications. PLoS ONE 14(2):e0211582.