Flow Cytometry Core
Sample Preparation
What do you need to bring for flow cytometry?
SAMPLE VOLUME, PREPARATION
Samples must be provided in a 12 x 75 mm 5 ml polystyrene tube (Fisher brand # 14-9563A, 14-9561D, or Falcon #352008, 352235; with the cap, 352054 or equivalent). It is the responsibility of the investigator to make sure the cells are aggregate-free. Samples can be filtered through a strainer cap (5ml Polystyrene Round-Bottom Tube with Cell-Strainer Cap, 35 micron, Falcon catalog number 35-2235) to remove aggregates. This is extremely important since large clumps of cells can clog up the fluidics of the flow cytometer which is going to take some effort to clean or worse damage the flow cell tip, which is costly to replace and will cause flow cytometry facility downtime. The filter cap tubes are pretty expensive, so if you are on a budget you can buy 30-50 micron nylon mesh in huge quantities and use that to filter your samples. You can also add 0.5 mM EDTA or 100-200 U/ml DNase to your buffer solution to prevent clogging.
The recommended cell concentration is 1 x 106/ sample, but 5 x 105/sample is the minimum required. Cells should be suspended in a volume of 0.25 ml – 1.0 ml.
In case of live cells a viability dye should be used to exclude dead cells with higher autofluorescence from the analysis. Depending on the fluorescent markers used, either propidium iodide (PI), 7AAD or DAPI may be added to the cells just prior to analysis. Only dead cells will take up these dyes. A very small amount of dye may be used to check viability, and will not be detrimental to the cells. As little as 0.5 - 1.0 ug/ml of either dye may be added immediately before the start of acquisition. If your sample is fixed, these dyes will not work. You will need a fixable live/dead marker, such as the Fixable Viability reagents from BD or one of the zombie reagents from Biolegend.
For Miltenyi autoMACS separation the cells should be in a single cell suspension. Pre-separation filters (such as Miltenyi 130-041-407) can be used to remove cell clumps. For up to 108cells, resuspend in 500ml PBS, 2mM EDTA, 0.5% BSA prior to sorting.
WAYS TO REDUCE BACKGROUND FLUORESCENCE
Reduction of background fluorescence enables clearer resolution of cellular subpopulations. However, with appropriate logical analysis this can be reduced. All samples generate an inherent degree of fluorescence (so called autofluorescence). Generally, this autofluorescence is increased with larger cells and is decreased at longer wavelengths. These larger cells include; monocytes, granulocytes and adherent cell lines. Moreover, these cells tend to form aggregates, binding predominantly through their Fc receptors. In general, if cells are visible with the naked eye, then its likely aggregates are present. These can be dispersed using: vortexing, EDTA (a little in the wash buffer may help), DNAse (used on unfixed cells) or filtering through a nylon mesh. If positive cells are sparse, then I recommend only using the first two. In addition, antibody conjugates can produce a low level of non-specific binding. When non-specific binding exceeds the level of autofluorescence then it can be difficult to resolve cells of interest. This can be caused by IgG isotypes that bind to the Fc receptor expressed on the cell surface, as well as by tandem conjugated-fluorochromes.
The type of media used to process experimental preparations can also significantly influence background fluorescence, phenyl red in particular is guilty of this. Experimental methods include, adding serum from the host, adding protein such as bovine serum albumin (BSA) or fetal bovine serum (FCS). These will reduce the level of background staining by blocking non-specific interactions, use of an appropriate wash buffer can also remove unbound antibody (wash buffer; 2% FCS, 0.1% BSA and 0.1% NaN3). Moreover, excessive processing of preparations can increase background fluorescence. The age and health of cells can also significantly contribute to background fluorescence. Furthermore, formaldehyde is common as a fixative in immunofluorescent experiments, although in older preparations the pH can drift and so influence fluorescence.
CONTROLS
I know it is painful, but as everywhere else in science you end up having a million control tubes for the two “real experiment” samples. Sorry about this, but see the bright side, this way your data will be both meaningful and publishable.
- Unstained ControlAn Unstained Control is used to detect "auto-fluorescence" or background staining of the cells of interest. Auto-fluorescence can be a significant problem, particularly in systems that contain monocytes/macrophages, cultured cells, or activated cells. Dead cells also often fluoresce right about where eGFP/FITC does (Ever wondered why are all the GFP positive transfected cells in your culture the ones that are dead?) It might be the other way around- they are really only green because they are dead) You will need this control even if compensation beads are used for comp control.
- Isotype ControlAn Isotype Control (i.e., where an antibody is used that has the same immunoglobulin subclass like IgG1, IgG2, as the test antibody, but a different specificity which is known to be irrelevant to the sample being analyzed) is needed to determine whether fluorescence that is observed is due to non-specific (e.g., due to Fc receptor on cells of interest) binding of the fluorescent antibody. Efforts should be made to use the same concentration of isotype antibody as the antibody of interest. Isotype controls have a long history, and hence are often applied without much afterthought. To be honest, isotype controls are hit-or-miss. Even if you are careful to use a nonspecific IgG2a as a control for your specific IgG2a antibody they still may have differences in terms of background staining, aggregation, etc. So, should you include isotype control? Read IsotypecontrolsTimetoletgo.pdf and Isotypeeditorial.pdf.
- Single Stains- to set compensation in a multi color experimentIn an ideal world fluorochromes would emit light in a narrow range, in the real one there is substantial overlap between spectra. Single stain controls is necessary to calculate and subtract spectral overlap between fluorochromes. You can use any cells for your single stained controls as long as you follow this rule: The positive and negative population for each single stain (i.e., PE+ and PE–) MUST have the same autofluorescence. If you have few cells and cannot spare enough for these controls, you can use antibody-capture beads.Any reagent can serve to stain your single stained control, provided it is labeled with the same fluorochrome as your experimental sample. In addition, the single stains should be at least as bright as the experimental sample. For example, you might want to substitute a CD8 PE for a CD34 PE.If you use a dye for dead/live discrimination (such as PI or 7AAD) you won't necessarily need a control for that since all positive events in this regard will be excluded from analysis.Caveats:Just because a number of fluorochromes are detected with the same detector, it does not mean you can use them interchangeably when setting up your compensation. They are all unique fluorochromes and have different spillover into other channels.Tandem dyes (such as Cy7PE, Cy7APC etc.) are manufactured by linking two fluorochromes. The amount of spillover into other colors depends on how they are manufactured. You cannot assume that all Cy7PE reagents are the same. Therefore you cannot substitute tandem dye reagents.You may ask: do I need to bring these controls every time? The answer is pretty much. Although we are often able to use the instrument settings from previous samples that were similar, over time the settings need adjustment.
- FMO (Fluorescence Minus One) – to set gatesIn multicolor experiments, it is not possible to set gates based on an entirely unstained or fully isotype stained control. FMO control leave out one reagent at a time. You may ask, I just gave you all the single stained compensation controls, why do you want even more? Well, spectral overlaps cause actually more than one trouble. There is the issue of compensation that we took care of in point 3. Then there is the issue that the overlap introduces extra variability into the spill over channels, hence making it more difficult to distinguish dim populations from the negative populations. The FMO control comes handy in this case. Can you ignore this? One school of thought is that the FMO controls are really to be used on an "as needed" basis. If you have a "hard to gate" channel that is a highly critical measurement for your study, than that is a candidate for an FMO control. Then you should run an EXACT match to that sample with all the fluors except the "hard to gate" one included. Then this FMO sample would be exceptionally useful.
Special notes for Annexin V Apoptosis Assay. Of the various combinations of fluorophores my favorite is DAPI for DNA dye with Annexin V -APC. Both are bright and don’t mess with each other. Also, collect everything! To harvest cell, you’ll remove the media. Put all of this in a 15 ml tube. Then you rinse your well, probably with PBS, add this PBS after the rinse to the old media in the 15 ml tube. Then you add EDTA, Trypsin-EDTA, versine or whatever your cells need to get detached into the wells. Once the cells are lose, add this to the same 15 ml tube. Dying cells are often detached or in the process of getting detached. If you don’t collect everything, you’ll under estimate apoptosis. Finally, the way how this assay goes south is lack of Ca++ ions. Annexin V needs that to bind to PS. So, make sure that you incubate the Annexin V with the cells in Ca++ containing binding buffer and if you wash the cells after, do it with Ca++ containing binding buffer.
Special note for PI cell cycle assay. This assay is the friend of those who are not in a rush. The alcohol fixation step likes to go long. An hour or two is probably not going to go well, I’d do minimum overnight. Heck, go on vacation and fix for 10 days. Make sure that you are stirring the sample when you are adding the alcohol to avoid clumping your cells up. Also, after the fixation the cells will not pellet as well as they did before. Spin them harder and/or longer than you do for cell culturing. RNAse treatment is really important. Without a good RNA digest PI cell cycle will look like the illustration from the Little Prince of the snake that swallowed an elephant, not a proper cell cycle. Longer is better than shorter. These samples are remarkably stabile. Leaving them on the bench I kept re-testing them and it took many days before the graphs started to look worse.
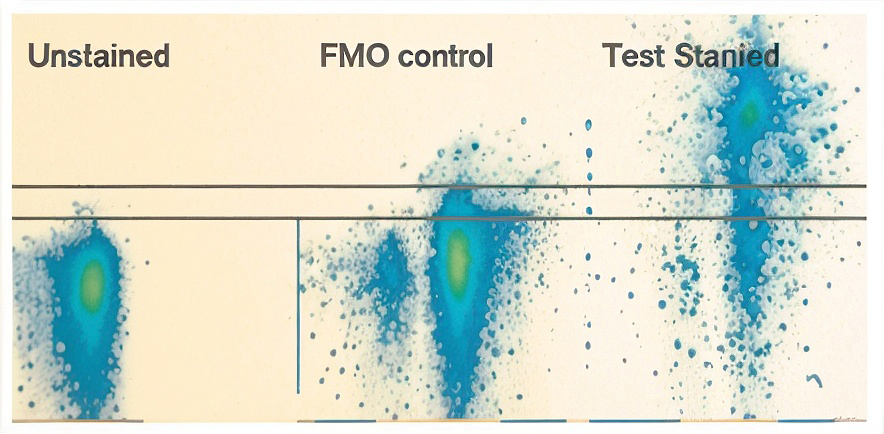
- FMO (Fluorescence Minus One) – to set gates
In multicolor experiments, it is not possible to set gates based on an entirely unstained or fully isotype stained control. FMO control leave out one reagent at a time. You may ask, I just gave you all the single stained compensation controls, why do you want even more? Well, spectral overlaps cause actually more than one trouble. There is the issue of compensation that we took care of in point 3. Then there is the issue that the overlap introduces extra variability into the spill over channels, hence making it more difficult to distinguish dim populations from the negative populations. The FMO control comes handy in this case. Can you ignore this? One school of thought is that the FMO controls are really to be used on an "as needed" basis. If you have a "hard to gate" channel that is a highly critical measurement for your study, than that is a candidate for an FMO control. Then you should run an EXACT match to that sample with all the fluors except the "hard to gate" one included. Then this FMO sample would be exceptionally useful.
From Perfetto SP, Chattopadhyay PK, Roederer M Seventeen-colour flow cytometry: unraveling the immune system. Nat Rev Immunol. 2004 Aug;4(8):648-5 Compensated, FITC, PE, Cy5PE, Cy7PE stained sample tested for PE staining using no primary v. FMO control to set gate on PE positive population. - EXPERIMENTAL Controls – scientific question
Experimental controls help you answer the scientific question being asked. They are NOT meant to set the compensation. Experimental controls allow a comparison of the sample from one experimental condition to another. For instance, if you are studying a cell line that has been drug treated, then you need to compare it to the same cell line that has not undergone treatment (sham treated).
Example staining setup of a 3 color experiment:
| Tube# | Description | FL1 | FL2 | FL3 |
|---|---|---|---|---|
| 0 | Unstained Control | - | - | - |
| 1 | Experimental Sample | CD3 FITC | CD4 PE | CD8 Cy5PE |
| 2 | Compensation Controls(Single stains – one for each fluorochrome used in the experiment) | CD3 FITC | - | - |
| 3 | - | CD4 PE | - | |
| 4 | - | - | CD8 Cy5PE | |
| 5 | Gating Controls (FMO – leave out one fluorochrome at a time) | - | CD4 PE | CD8 Cy5PE |
| 6 | CD3 FITC | CD4 PE | CD8 Cy5PE | |
| 7 | CD3 FITC | - | - | |
| 8 | Experimental Controls (fully stain healthy or untreated samples to compare to experimental sample) | CD3 FITC | CD4 PE | CD8 Cy5PE |
| 9 | CD3 FITC | CD4 PE | CD8 Cy5PE | |
| 10 | CD3 FITC | CD4 PE | CD8 Cy5PE | |
* no stain added or add isotype matched control stain.